Yonsei University researchers have conducted a comprehensive functional analysis of the ataxia telangiectasia mutated (ATM) gene, which is associated with a high risk of cancer and is closely linked to the development of rare diseases, Severance Hospital said on Tuesday.

Professor Kim Hyong-bum of the Department of Pharmacology at Yonsei University College of Medicine, along with Instructor Lee Kwang-seob and Student Min Joon-goo of the Graduate School of Medical Science, Yonsei University College of Medicine, announced that they successfully evaluated the functions of all 27,513 possible single-nucleotide variants (SNVs) in the ATM gene.
The findings were recently published in the latest issue of the international journal Cell (impact factor 42.5).
ATM plays a critical role in detecting and repairing DNA damage in the body. When the gene does not function properly, the risk of developing cancers such as breast, colon, and pancreatic cancer increases significantly, and the prognosis for affected patients is often poor. Mutations in ATM can also lead to rare diseases, including ataxia telangiectasia.
Identifying variants that impair ATM function is crucial for predicting cancer risk in healthy individuals carrying the mutation, as well as for assessing the prognosis of cancer patients.
While recent advances in genomic analysis have improved the precision of diagnosing genetic diseases and cancers, many genetic variants remain of unknown significance. As a result, they cannot yet be reliably used for patient diagnosis or treatment. This challenge is particularly pronounced for the ATM gene, which contains approximately 9,000 protein sequences and a large number of variants -- too complex to be fully assessed using traditional statistical methods.
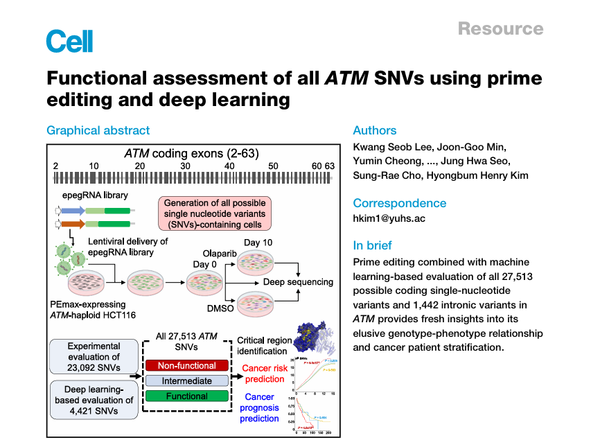
The team analyzed all 27,513 SNVs in the entire protein-coding region of ATM (62 exons). Of these, functions of 23,092 variants were directly confirmed in cell experiments using the latest gene-editing technology called “prime editing,” while the remaining 4,421 variants, which were difficult to evaluate experimentally, were predicted for their effects on cell survival using an AI model called DeepATM.
By analyzing the effect of each variant on cell survival, the team was able to distinguish, with high accuracy, between variants that are detrimental to the function of the gene and those that are not.
Using genomic and clinical data from nearly 500,000 individuals in the UK Biobank, the team found that individuals with the deleterious variants identified in the study had an approximately 1.4-fold increased risk of developing cancer compared to those without the deleterious variants. They also demonstrated greater than 95 percent agreement with data from ClinVar, an international database of genetic variants. When existing cancer genomic data (cBioPortal) was reinterpreted in light of this study, it also confirmed that the survival rate of cancer patients varies depending on whether they carry a deleterious variant of ATM.
“This study is significant because it provides the possibility of accurately identifying variants in the ATM gene, which is difficult to interpret, on a large scale,” said Professor Kim. “In the future, similar analyses will be possible for other genes, which will contribute to the development of genome-based precision medicine.”
Related articles
- Yonsei Cancer Center study identifies MET gene as key target across solid tumors
- Simple nasal swab identifies type 2 chronic rhinosinusitis with high accuracy
- 4-day workweek pilot at Severance shows gains in nurse retention, care quality
- Bill seeks to block overseas transfer of sensitive genomic and biometric data